Edwardsi haigus. Edwardsi sündroom - põhjused, sümptomid, diagnoos ja ravi. Sündroomi kõrvalekalded
Enamik naisi teab hästi, mis on Downi sündroom. Ja raseduse ajal õpivad paljud seda väga harva, kuid on veel üks kromosomaalne häire, mida nimetatakse Edwardsi sündroomiks. Ja paljud on mures selle pärast, kuidas teada saada, kui suur on Edwardsi sündroomiga lapse saamise oht ja kuidas sellist patoloogiat raseduse ajal diagnoositakse?
Mis on Edwardsi sündroom?
Edwardsi sündroom on geneetiline haigus, mida iseloomustab VIII kromosoomi dubleerimine (trisoomia) ja mis avaldub mitmete iseloomulike väärarengutena lootel raseduse ajal, mis sageli põhjustab lapse surma või tema puude. See tähendab, et lapsel moodustub 46 kromosoomi asemel 47, see lisakromosoom annab haigusele teise nime - trisoomia 18. Sündroom sai nime teadlase John Edwardsi järgi, kes kirjeldas seda esmakordselt 1960. aastal.
Miks tekib Edwardsi sündroom - patoloogia põhjused
Isegi kui vanemad on terved ja perekonna ajaloos sellist patoloogiat pole, võib 18. kromosoomiga laps sündida igal naisel.
Teatavasti on igas inimese rakus 46 kromosoomi ning naise ja mehe sugurakkudes kummaski 23 kromosoomi, mis munaraku viljastamise käigus kombineerituna annavad samuti kokku 46 kromosoomi. Kui me räägime Edwardsi sündroomist, siis selle välimuse põhjused pole teada.
Praegu on teada vaid see, et teatud geneetiliste mutatsioonide tulemusena tekib 47. lisakromosoom (täiendav kromosoom 18. kromosoomipaaris, millest saab seega mitte 2, vaid 3).
95% kõigist Edwardsi sündroomi väljakujunemise juhtudest on see rakkudes 18. lisakromosoom (trisoomia), kuid 2% puhul esineb ainult 18. kromosoomi "pikenemine" (translokatsioon), kui kromosoomid jäävad normaalseks ja on 46.
3% Edwardsi sündroomi juhtudest tekib "mosaiiktrisoomia", kui kehas leitakse täiendav 47. kromosoom mitte kõigis rakkudes, vaid ainult teatud osas. Kliiniliselt kulgevad kõik kolm Edwardsi sündroomi varianti peaaegu ühtemoodi, kuid esimene variant võib erineda haiguse raskema kulgemise poolest.
Kui levinud see patoloogia on?
Edwardsi sündroomiga lapsed surevad emakasse ligikaudu 60% juhtudest. Sellele vaatamata on geneetiliste haiguste hulgas see sündroom ellujäänud imikute puhul üsna levinud, esinemissageduselt teisel kohal Downi sündroomi järel. Edwardsi sündroomi levimus on 1 juhtum 3-8 tuhandest lapsest.
Arvatakse, et tüdrukutel esineb seda haigust 3 korda sagedamini ja Edwardsi sündroomi risk suureneb oluliselt, kui rase naine on 30-aastane või vanem.
Edwardsi sündroomi suremus esimesel eluaastal on umbes 90% ja keskmine eluiga rasketel haigusjuhtudel poistel on 2-3 kuud ja tüdrukutel 10 kuud ning ainult vähesed jäävad täiskasvanuks. Kõige sagedamini surevad Edwardsi sündroomiga lapsed lämbumise, kopsupõletiku, südame-veresoonkonna puudulikkuse või soolesulguse tõttu – kaasasündinud väärarengute põhjustatud tüsistustesse.
Kuidas sündroom avaldub lapsel?
Edwardsi sündroomi sümptomid võib jagada mitmeks suureks rühmaks:
Esimesse rühma kuuluvad lapse välimusele iseloomulikud sümptomid:
- Sünnihetkel väike kehakaal (umbes 2100–2200 grammi)
- Ebaproportsionaalselt väike pea
- Ülemiste või alumiste lõualuude arenguhäired (mikrognaatia)
- Näo kuju moonutamine ja väära haardumise teke
- Suulaelõhe (suulaelõhe) või huulelõhe (huulelõhe)
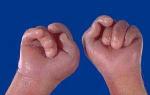
- Käe sõrmed on kokku surutud, rusikas ebaühtlased
- Madala asetusega kõrvad
- Alajäsemete sõrmede vöö või täielik sulandumine
- kaasasündinud lampjalgsus
- "Kiik jalg"
- Suhteliselt väike suulõhe (mikrostoomia)
Teise rühma kuuluvad siseorganite, motoorika ja neuropsüühilise arengu häirete tunnused:
- Kaasasündinud südamedefektide olemasolu, näiteks avatud foramen ovale, ventrikulaarne vaheseina defekt, avatud arterioosjuha jne.
- Kubeme- või nabasongide areng.
- Seedeorganid: gastroösofageaalne reflukshaigus, neelamis- ja imemisrefleksi rikkumine, söögitoru või päraku atreesia, Meckeli divertikulaar, soolestiku asukoha rikkumine.
- Kesknärvisüsteem: hiline neuropsüühiline areng, vaimne alaareng, väikeaju, kollakeha alaareng, ajukonvolutsioonide silumine või atroofia.
- Urogenitaalsüsteem: krüptorhidism, hüpospadiad poistel, kliitori hüpertroofia, tüdrukute munasarjade alaareng, sõltumata soost - hobuseraua või segmenteeritud neer, kusejuhade kahekordistumine.
- Strabismus, skolioos, lihaste atroofia.
Kuidas patoloogiat raseduse ajal teada saada - diagnoos
Patau sündroom, Edwardsi sündroom ja muud trisoomiad on kõige paremini tuvastatavad enne lapse sündi. Reeglina viiakse selle sündroomi sünnieelne diagnoos läbi kahes etapis:
- Perioodiks 11–13 nädalat (sõeluuringud, mis põhineb erinevatel biokeemilistel analüüsidel naisel).
- Loote karüotüübi määramine nende riskirühma rasedatel naistel.
11–13. nädalal määratakse naise veres teatud verevalkude tase: β-hCG (inimese koorionihormooni β-subühik) ja rasedusega seotud plasmavalk A. Seejärel arvutatakse neid andmeid arvesse võttes raseda vanus, Edwardsi sündroomiga lapse saamise risk ja moodustatakse rasedate naiste riskirühm.
Edasi võetakse riskirühmas hilisemal ajal lootelt materjali täpse diagnoosi panemiseks: 8–12 nädalal on koorionivilluse biopsia, 14–18 lootevee uuring (amniootilise vedeliku uuring), pärast 20. nädalat - kordotsentees (emakasisene vereproovide võtmine nabaväädi lootelt ultraheli juhtimisega). Pärast seda määratakse saadud materjalis KF-PCR (kvantitatiivne fluorestsentspolümeraasi ahelreaktsioon) abil täiendava 18. kromosoomi olemasolu või puudumine.
Kui rase naine ei ole läbinud geneetilist sõeluuringut, tehakse hiljem ultraheli abil Edwardsi sündroomi esialgne diagnoos. Muud kaudsed nähud, mille alusel saab hiljem kahtlustada Edwardsi sündroomi:
- Anomaaliate esinemine pea luude ja pehmete kudede arengus (“suulaelõhed”, madala asetusega kõrvad, “huulelõhed” jne).
- Kardiovaskulaarsüsteemi, urogenitaalsüsteemi, aga ka lihasluukonna defektide tuvastamine.
Sündroomi diagnostilised tunnused lapsel
Pärast lapse sündi on Edwardsi sündroomi esinemise peamised diagnostilised tunnused järgmised:

Iseloomuliku dermatograafilise pildi tunnused:
- arenemata distaalne paindevolt sõrmedel
- põiki peopesa soone olemasolu 1/3 juhtudest
- kaared sõrmeotstes
- muutus peopesa nahamustris: aksiaalse triraadiuse distaalne asukoht ja harja arvu suurenemine.
Edwardsi sündroom ultraheliuuringul - koroidpõimiku tsüstid
Varases staadiumis on Edwardsi sündroomi ultraheliga äärmiselt raske kahtlustada, kuid 12 rasedusnädalal ilmnevad juba sellele iseloomulikud kaudsed sümptomid. :
- Loote kasvupeetuse tunnused
- Bradükardia (loote südame löögisageduse langus)
- Omfalotseele (kõhuõõne songa olemasolu)
- Nina luude visualiseerimine puudub
- Nabanööris on üks arter, mitte 2
Ka ultraheliga saab tuvastada veresoonte põimiku tsüstid, mis on õõnsused, milles on vedelikku. Iseenesest nad tervisele ohtu ei kujuta ja kaovad 26. rasedusnädalaks. Sellised tsüstid kaasnevad aga väga sageli erinevate geneetiliste haigustega, näiteks Edwardsi sündroomiga (sel juhul leitakse tsüstid 1/3 selle patoloogia all kannatavatest lastest), mistõttu selliste tsüstide avastamisel suunab arst raseda edasi. naine geenikonsultatsioonile uuringuks.
Ravi
Kuna Edwardsi sündroomiga lapsed elavad harva kuni aasta, on ravi esmalt suunatud nende väärarengute parandamisele, mis on eluohtlikud:
- toidu läbipääsu taastamine soolestiku või päraku atreesias
- toitmine läbi sondi neelamis- ja imemisreflekside puudumisel
- antibakteriaalne ja põletikuvastane ravi kopsupõletiku korral
Suhteliselt soodsa haiguse kulgemise korral korrigeeritakse mõningaid kõrvalekaldeid ja väärarenguid: "suulaelõhe", südamerikete, kubeme- või nabasongi kirurgiline ravi, samuti sümptomaatiline medikamentoosne ravi (kõhukinnisuse lahtistite väljakirjutamine, vahusurutajad). kõhupuhitus jne). 
Edwardsi sündroomiga lapsed on altid sellistele haigustele nagu:
- keskkõrvapõletik
- neeruvähk (Wilmsi kasvaja)
- kopsupõletik
- konjunktiviit
- pulmonaalne hüpertensioon
- apnoe
- kõrge vererõhk
- frontiit, sinusiit
- kuseteede infektsioonid
Seetõttu hõlmab Edwardsi sündroomiga patsientide ravi nende haiguste õigeaegset diagnoosimist ja ravi.
Prognoos lapsele
Enamikul juhtudel on prognoos halb. Vähestel Edwardsi sündroomiga lastel, kes elavad täiskasvanueani, on tõsine vaimupuue ja nad vajavad pidevalt välist hoolt ja järelevalvet. Sobivate tegevustega suudavad nad aga reageerida mugavusele, naeratada, iseseisvalt süüa ja suhelda hooldajatega, omandades erinevaid oskusi ja võimeid.
Valdav enamus inimesi teab, et on olemas selline haigus nagu Downi sündroom. Keegi teab, et see on kromosomaalne haigus, ja keegi usub, et see on vaimne või neuroloogiline kõrvalekalle normist. Kuid vähesed teavad, et kromosomaalseid haigusi on palju rohkem ja üks neist on Edwardsi sündroom. Võib-olla on see tingitud Edwardsi sündroomiga sündinud laste lühikesest elueast ja selliste laste suhteliselt madalast osakaalust tervete laste hulgas (1 sündroomiga laps 7000 elussündi kohta ja 6000 vastsündinu kohta).
Teave Loodusliku raseduse katkemise protsent geneetiliselt defektse lootega ja abortide protsent pärast loote diagnoosimist on kõrge.
Mis see on?
Edwardsi sündroom on raske kromosomaalne häire, mida nimetatakse ka trisoomiaks 18. Selle haigusega kaasnevad kõigi elundite rasked patoloogiad ja mitmed väärarengud. Kõige sagedamini sünnivad tüdrukud Edwardsi sündroomiga: neid on kolm korda rohkem kui sama patoloogiaga poisse. Võib-olla on see tingitud asjaolust, et naissoost loodete loomulik elu toetav mehhanism “töötab” paremini.
Lisaks Austraalia arstiteadlased uurisid platsentasid ja jõudsid järeldusele, et neil on erinev struktuur ja nad täidavad oma kaitsefunktsioone erineval viisil, olenevalt raseda loote soost. Varem on statistikas märgitud, et väärarengutega tüdrukute tiinus on kõrgem kui sarnaste kõrvalekalletega poistel ning vastsündinud tüdrukute suremus on madalam kui poistel, kusjuures kõik muud näitajad on võrdsed.
Edwardsi sündroomi põhjused
Edwardsi sündroomi põhjuseks on täiendava 18. kromosoomi olemasolu sigooti karüotüübis. Tavaliselt on diploidsel komplektil kaks kromosoomi. Inimese kehas on kõik rakud diploidsed, see tähendab, et nende tuumas on kaks kromosoomi. Haploidne - tuumas on üks kromosoom - ainult sugurakud (sugurakud). Haploidsed sugurakud on tingitud asjaolust, et viljastamise ajal toob iga rakk oma 23 kromosoomi komplekti. Seega on lootel vajalikud 46 kromosoomi – pool kummaltki vanemalt.
Mõnikord juhtub viljastamise ajal rike ja moodustub vale arv kromosoome. See "ebakorrapärasus" lihtsalt toimib kromosomaalsete haiguste põhjusena: Edwardsi sündroom,. Kõik need haigused põhjustavad tõsiseid kõrvalekaldeid laste arengus, mõjutavad nende tervist ja vähendavad oluliselt patsientide elukvaliteeti. Kõik need lapsed, olenemata kõrvalekallete astmest, ei ole iseseisvaks eluks võimetud ja vajavad pidevat hoolt.
Teave Mõnikord on sugukromosoomide arv häiritud. See on paljude muude patoloogiate põhjus. Harva sisaldab sugurakk juba vale arvu kromosoome. Sellised haigused põhjustavad seksuaalset infantilismi, võimetust seksida, viljatust, kuid üldiselt ei avalda nad patsientide üldisele tervisele nii katastroofilist mõju ja võimaldavad neil elada peaaegu täisväärtuslikku elu.
Kõik kromosoomianomaaliad on seotud ema vanusega esmasünnituse ajal. Arvatakse, et 45 aasta pärast on naisel võimalus sünnitada kõnealuse patoloogiaga laps, mis võrdub 0,7%. Edwardsi sündroomiga lapse sünnitanud 20-30-aastaste noorte emade arv on aga suurem kui Downi sündroomiga ja Patau sündroomiga lapsi sünnitanud samaealiste emade arv. Ema vanus ei ole nii selgelt seotud sellise kromosomaalse patoloogiaga nagu Edwardsi sündroom. Arvatavasti on täiskasvanueas eostatud laste geneetiliste kõrvalekallete risk seotud võimalike geenimutatsioonidega ema rakkudes elu jooksul ebasoodsate keskkonnategurite mõjul. See on aga vaid oletus ega seleta kõiki kromosoomihaigustega laste sünnijuhtumeid.

Haiguse tunnused
Edwardsi sündroomiga sündinud laste diagnoosimine toimub peamiselt välimuse järgi. Mis tahes kromosoomianomaaliaga vastsündinud näevad välja spetsiifilised. Igal haigusel on oma fenotüübilised tunnused. Normaalse ja isegi ülemäärase rasedusega imikud sünnivad väikese kaaluga - ligikaudu 2200 g. Tõenäoliselt on selle põhjuseks imiku kudede vereringe- ja troofilised häired, aga ka nabanööri toitumisvaegus kogu raseduse vältel.
Haiguse fenotüübilised ilmingud on väga mitmekesised:
- kolju ja näo luude anomaaliad;
- ajukolju on tavaliselt dolichocephalic – pika ja kitsa kujuga;
- alalõug on vähearenenud, suuava ebakorrapärane ja väike;
- silmalõhed kitsad ja lühikesed, ebakorrapärase kujuga kõrvad puuduvate osadega ja madala asetusega;
- mõnikord puudub kuulmekäik või on see väga lühike, mille tõttu sünnivad lapsed kurdina;
- rinnaku on lühike, lai, roietevahelised ruumid on oluliselt vähenenud;
- jalg on tavaliselt deformeerunud - suur varvas on paks ja lühike, kand eendub järsult, kaar langeb.
Võimalikud on ka muud mittespetsiifilised ilmingud - polüdaktsus, oligodaktilisus ja muud kõrvalekalded. Mittespetsiifilisus tähendab, et need kõrvalekalded esinevad teiste geneetiliste häirete korral.
Edwardsi sündroomiga üksikasjaliku morfoloogilise uuringu käigus avastavad vastsündinu südame ja suurte veresoonte väärarengud, teiste siseorganite patoloogiad. Ilma sekkumiseta ei pääse toit sageli seedekulglast läbi ning soolte ja põie tühjendamise võime halveneb.
Teave Kõigil Edwardsi sündroomiga patsientidel on väikeaju hüpoplaasia (selle tõttu on lapse liigutused koordineerimata ja kontrollimatud, mis püsib kogu eluperioodi jooksul), kollakeha hüpoplaasia (põhjustab krampe, sensoorsete reaktsioonide häireid, termoregulatsiooni võimetust, madal nutmise modulatsioon), oliivide struktuuride rikkumine - pikliku medulla osad (selle tõttu on probleeme spontaanse hingamise ja vereringega, eriti hingamisseiskus on üks levinumaid surmapõhjuseid lastel Edwardsi sündroom).
Edwardsi sündroomiga lastel on ka väljendunud lihasnõrkus, kõigi lihaste ebanormaalne lõdvestus, ei reageeri stiimulitele – valgus, heli, puudutus, refleksid ei arene, mõnikord isegi imemine ja neelamine. Üheaastase ja vanemate laste puhul, mida juhtub üliharva, peaaegu mitte kunagi, on sügav vaimne alaareng kuni idiootsuseni ja täielik intelligentsuse puudumine.
Edwardsi tõbi ultraheliuuringul
Kõik rasedad naised peavad läbima sõeluuringu loote kromosoomianomaaliate esinemise suhtes. Edwardsi sündroomi diagnoositakse mitte mingil moel kuni 12. nädalani, kuigi munaraku viljastumise hetkest on kõrvalekaldeid, kuid alates 12. nädalast on võimalik tuvastada sellele haigusele iseloomulikke sümptomeid. Need sisaldavad:
- bradükardia (loote südame löögisageduse rikkumine südame löögisageduse languse kujul),
- omphalocele (song kõhuõõnes),
- visuaalselt määratletud nina luude puudumine,
- ühe arteri puudumine nabanööris (tavaliselt peaks nabanööris olema kaks arterit),
- Veresoonte põimiku tsüsti olemasolu (tsüst ise ei kujuta endast ohtu lootele ja kaob spontaanselt 26. rasedusnädalaks, kuid esineb sageli loote erinevate geneetiliste haiguste, sh Edwardsi sündroomi taustal).
Vähemalt ühe kõrvalekalde olemasolu on põhjus raseda täiendavale uuringule saatmiseks. Naise sunniviisilist läbivaatust ei toimu, ta peab andma vabatahtliku nõusoleku kõikideks diagnostilisteks protseduurideks ja manipulatsioonideks. Arst on kohustatud sel juhul selgitama diagnostiliste protseduuride tähendust ja tähtsust, samuti nende läbiviimise korda.
Rohkem võib tuvastada neuraaltoru defektist tingitud loote spina bifida, vedeliku suurenemist krae ruumis, luude lühenemist, kolju deformatsiooni ja muutusi aju struktuurides. Need kõrvalekalded on iseloomulikud paljudele kromosoomianomaaliatele.
Ultraheli loote kromosomaalsete haiguste tuvastamiseks kutsuvad ja viivad läbi arstid, kes on saanud eriväljaõppe geneetiliste kõrvalekallete valdkonnas. Selleks saadetakse naine tavaliselt teise asutusse uuringutele, kuna igal polikliinikul pole vajalikku varustust ja vastava väljaõppega spetsialisti.
Lisaks Rasedate 1. trimestri sõeluuringu programm sisaldab ka vereseerumi analüüsi, sest mitte ükski kõige täpsem ultraheliaparaat ei suuda absoluutse täpsusega näidata loote haiguste esinemist või puudumist.
Kromosomaalse haigusega lapse saamise risk arvutatakse järgmiste näitajate järgi:
- rasedusega seotud plasmavalgu A kogus (seerumianalüüs)
- inimese kooriongonadotropiini vaba β-subühiku kogus (seerumianalüüs),
- morfoloogilised tunnused (ultraheli).
Individuaalne risk saada geneetilise arenguanomaaliaga laps on 1/100 või rohkem.
Kõige täpsema diagnoosi saab teha lootematerjali uuringu põhjal.
Selle jaoks . Milline protseduur viiakse läbi, sõltub rasedusajast:
- 8-12 nädala jooksul (koorioni koel on sama kromosoomide komplekt kui lootel, seetõttu näitab see täpselt geneetiliste kõrvalekallete olemasolu või puudumist,
- 14-18 nädalal (amnionivedeliku kogumine ja analüüs),
- 18-20 nädalal ja hiljem (nabaväädivere kogumine ja analüüs).
Amniotsentees ja kordotsentees tehakse sageli sama diagnostilise protseduuri käigus koos. See võimaldab saada põhjalikumaid ja täpsemaid tulemusi.
Tähtis Kromosomaalsete häirete, sealhulgas loote Edwardsi sündroomi ja lapse elule ja tervisele ebasoodsa prognoosiga kaasasündinud väärarengute diagnoosimisel tehakse perinataalkonsiiliumi otsusel ja raseda vabatahtlikul nõusolekul abort. teostatakse sõltumata gestatsiooni vanusest.
Naist tuleb teavitada loote diagnoosist, diagnoositud haiguse käigust, sellise anomaaliaga sündinud laste arengu iseärasustest ja elukvaliteedist.
Pärast 16-18 rasedusnädalat tehakse loote kromosoomianomaaliate kolmekordne sõeltest. Biokeemilise vereanalüüsi käigus hinnatakse järgmisi näitajaid:
- α-fetoproteiin (alfa-FP);
- inimese koorionihormooni vaba β-subühik (beeta-hCG);
- estrioolivaba.
Sündroomi tüübid
Esineb Edwardsi sündroomi täieliku trisoomia tüüp, kui lapse keha kõigis rakkudes on täiendav kromosoom (see on 95% kõigist juhtudest), mosaiiktrisoomia, kui kõigis rakkudes ei leidu täiendavat kromosoomi (3%). kõigist juhtudest), Edwardsi sündroomi osalise trisoomia tüüp, kui osa kromosoomist on seotud teise kromosoomiga (2% kõigist juhtudest). Trisoomia 18 esimene ja teine variant arenevad enne viljastumist ühes vanemate sugurakkudest. Kolmas variant areneb pärast viljastamist.
Edwardsi sündroomi teise ja kolmanda arengutüübi korral on need veidi vähem väljendunud, kuid ei võimalda ka lapsel täisväärtuslikku elu elada.
Edwardsi sündroomi kulg
Kuna Edwardsi sündroomi seostatakse siseorganite arengus mitmete patoloogiatega, on tema elu prognoos äärmiselt ebasoodne. Esimese kolme kuu jooksul pärast sündi sureb umbes 60% lastest. Umbes 5-10% elab aastani ja alla 1% noorukieani. Need lapsed on sügavad oligofreenikud. Nad ei räägi, ei saa kõnekõnest praktiliselt aru (nende hulgas on sageli kurte), ei mäleta midagi, ei koordineeri oma tegevust, ei kontrolli tungi urineerida ja roojata, ei saa iseseisvalt süüa (vastavalt mõned teated, mõned noorukid on õppinud iseseisvat söömist ilma abita). söögiriistad), Edwardsi sündroomiga lapsed praktiliselt ei kõnni ega kõnni suurte raskuste ja toega. Neil on järsult vähenenud tundlikkus, sealhulgas valu. Enamasti on neil raske oma keha asendit muuta. Enamikule Edwardsi sündroomiga patsientidest ei ole sisukas tegevus kättesaadav. Nad ei erista inimesi üksteisest, nende tähelepanu pole peaaegu kunagi keskendunud. Nad ei saa nutta ega naerda, kuid on olnud juhtumeid, kui seda haigust põdevad lapsed suutsid naeratada ja reageerisid isegi kiindumusele positiivselt.
Edwardsi sündroomiga laste kõige levinumad surmapõhjused on hingamisseiskus või südameseiskus.
Edwardsi sündroomi ravi ja tagajärjed
Kromosomaalsete häirete korrigeerimine ja ravi on võimatu. Peaaegu kogu Edwardsi sündroomiga laste keha arengu kõige tugevamate patoloogiate tõttu on kirurgiline ravi siseorganite struktuuride ja toimimise taastamiseks peaaegu ebaefektiivne. Kogu arstiabi taandub pidevale lapse elu jälgimisele ja tema pere toetamisele.
Vahetult pärast sündi on ravi suunatud nende defektide parandamisele, mis kujutavad endast kõige ilmsemat ohtu lapse elule: söögitoru ja/või soolte arengu anomaaliate korral taastatakse kirurgiliselt toidu läbimine, toitmine läbi sondi. on välja kujunenud, kuna imemis- ja neelamisrefleksid sageli puuduvad või on väga nõrgalt väljendunud. Vajadusel viiakse läbi põletikuvastane ja antibakteriaalne ravi.
Teave Kui beebi prognoos on suhteliselt soodne, võetakse meetmeid südame- ja veresoonkonna defektide kirurgiliseks korrigeerimiseks, herniad kõrvaldatakse ja suulae täielikult või osaliselt elimineeritakse (). Samuti on ette nähtud ravimid väljaheite stimuleerimiseks ja soolestiku gaaside eemaldamiseks, kuna need protsessid on soolepatoloogiate tõttu keerulised.
Lapsed kannatavad kogu elu põsekoopapõletiku, konjunktiviidi, keskkõrvapõletiku, kopsupõletiku, urogenitaalsüsteemi infektsioonide all. Sageli areneb neil neeruvähk. Sellistel juhtudel on ravi sümptomaatiline.
Edwardsi sündroom on üks geneetilistest haigustest, mis tulenevad kromosoomide struktuuri rikkumisest. Lootel on dubleeritud 18. kromosoom, mille tulemusena on tema arengus terve kompleks defekte. Kavandatud 46 kromosoomi asemel areneb lapsel 47.
Edwardsi sündroom sai nime teadlase John Edwardsi järgi, kes kirjeldas seda esimest korda eelmise sajandi 60ndatel.
Tuleb märkida, et sündroomiga laps võib sündida isegi täiesti tervetel vanematel, mistõttu haiguse peamised põhjused on siiani teadmata. Ainult üks on selge, et patoloogia tekib geneetiliste mutatsioonide tagajärjel ja võib areneda kolmes peamises vormis:
- Trisoomia (95% juhtudest);
- Translokatsioon (2% juhtudest);
- Mosaiiktrisoomia (3% juhtudest).
Kõigi kolme patoloogia vormi kliinilised ilmingud on absoluutselt identsed, kuid trisoomia on reeglina raskem.
Sümptomid
Edwardsi sündroomi tekkega rasedal naisel sureb 60% juhtudest loode isegi emakas. Kuid juhtub ka seda, et imikud jäävad ellu. Patoloogia levimusjuhtumid on ligikaudu 1 beebil 5000-st. Tüdrukutel esineb haigus kolm korda sagedamini. Arvatakse, et vanematel lapseootel emadel on selle arengu oht suurem.
Tavaliselt avastatakse Edwardsi sündroomi sümptomid ultraheliuuringu etapis, mis viiakse läbi raseduse ajal. Juba 12 nädala pärast võib avastada patoloogiale iseloomulikke tunnuseid. Eriti:
- loote kasvupeetuse nähud;
- südame löögisageduse vähenemine;
- kõhukelme hernia;
- nina luude puudumine;
- üks arter nabanööris kahe asemel.
Lisaks võib sündroomi arengu sümptom rasedatel olla veresoonte põimiku tsüstid. See tähendab, õõnsused vedelikuga. Need ei kujuta ohtu lapseootel emale ega lootele ning kaovad sageli 26. rasedusnädalaks. Kuid just seda tüüpi tsüstid on loote erinevate geneetiliste kõrvalekallete peamine märk. Need ilmnevad kolmandikul patoloogiaga lastest, nii et arst saadab tsüstide avastamisel tulevase ema täiendavale geneetilisele uuringule.
Edwardsi sündroomi diagnoosimine raseduse ajal
Edwardsi sündroom on määratletud kahes peamises etapis:
- kui läbite sõeluuringu 11–13 rasedusnädala jooksul;
- loote karüotüübi määramisel.
Esimesel etapil läbib rase naine laboratoorse diagnostika. Pärast kõiki analüüse, võttes arvesse saadud andmeid ja tulevase ema vanust, saab arst arvutada geneetiliste kõrvalekalletega lapse saamise riski.
Kui rase naine on ohus, võetakse lootelt hiljem materjal, mille abil pannakse paika täpne diagnoos. 14.–18. nädalal tehakse lootevee uuring, mille käigus uuritakse lootevett, seejärel raseduse lõpuks kordotsentees. Selle uuringu käigus võetakse loote nabanöörist verd ja seejärel otsitakse rakkudes lisakromosoomi olemasolu.
Samuti saab ultraheli abil uurida rasedat naist. Sellise läbivaatuse käigus avastatakse lapse luu-lihaskonna arengu anomaaliad, tema kolju luud. Samuti leitakse urogenitaal- ja kardiovaskulaarsüsteemi väärarenguid.
Kõik need diagnostikameetodid on väga informatiivsed ja umbes 90% täpsed.
Tüsistused
Patoloogia prognoosi võib nimetada pettumuseks. Edwardsi sündroomiga sündinud imikute suremus on esimesel eluaastal umbes 95%. Surm saabub eluga kokkusobimatute häirete tõttu. Hingamissüsteemi organite ja südame-veresoonkonna süsteemi arengus võivad tekkida defektid. Imikud, kellel õnnestus ellu jääda, kannatavad tavaliselt vaimse alaarengu raske vormi all.
Tulevane ema, kes on selliste kõrvalekalletega lapse sünniks valmis, nõuab talle suuremat tähelepanu ja kontrolli tema seisundi üle. Sellised lapsed jäävad füüsilises ja vaimses arengus maha. Nõuetekohase hoolduse korral saab laps õppida iseseisvalt sööma ja pead tõstma.
Ravi
Mida sa teha saad
Lapseootel ema peaks mõistma, et kui diagnostika tulemused näitasid Edwardsi sündroomi esinemist lootel, jääb diagnoos lapsele kogu eluks. Seetõttu on tema enda otsustada, kas ta on valmis sellise vastutuse enda peale võtma või on ainuõige variant rasedus varases staadiumis katkestada.
Mida teeb arst
Edwardsi sündroom on geneetiline häire ja see võib erineval määral mõjutada lapse keharakke. See tähendab, et patoloogia ravimiseks on vaja "parandada" kromosoomide struktuuri rikkumisi. Praegu on see võimatu, mistõttu haigus liigitatakse ravimatuks. Kuigi tulevikus on arstide ja teadlaste hinnangul siiski võimalik leida universaalne ravim teistsuguse iseloomuga kromosoomihäiretele.
Kuna haigus on ravimatu, piirdub ravi toetavate meetmetega. Arst püüab tugevdada lapseootel ema moraali, annab talle juhiseid sellise patoloogiaga beebi edaspidiseks kasvatamiseks ja hooldamiseks. Kui geneetiline kõrvalekalle avastatakse raseduse alguses, soovitab arst tavaliselt selle katkestada. Mida teha - vanemad otsustavad. Kui nad on valmis võtma vastutuse Edwardsi sündroomiga beebi kasvatamise eest, peaksid nad olema valmis selleks, et sellised lapsed elavad harva kuni aasta ning neil on alati süsteemide ja organite töös ja struktuuris mingeid defekte.
Ärahoidmine
Sündroomi arengu vältimiseks raseduse ajal ei ole ennetavaid meetmeid. Ainus ennetusmeede on suunatud geneetilise häire varajasele avastamisele, seetõttu tehakse igat tüüpi diagnostikat tavaliselt raseduse 1-2 trimestril.
Edwardsi sündroom on kromosoomianomaaliatest põhjustatud haigus, millega kaasneb terve kompleks erinevaid kõrvalekaldeid ja arenguhäireid. Selle haiguse põhjuseks on 18. kromosoomi trisoomia, see tähendab, et kehas on kromosoomi lisakoopia, mis põhjustab erinevaid geneetilist laadi tüsistusi.
Edwardsi sündroomiga lapse saamise keskmine risk on 1 5000-st ja enamik selle sündroomiga vastsündinuid sureb esimeste elunädalate jooksul. Vähem kui 10% elab ühe aasta. Edwardsi sündroom tähendab sügavat vaimset alaarengut ja arvukaid kaasasündinud väärarenguid, mis mõjutavad nii välis- kui ka siseorganeid. Kõige levinumad on süda, aju, neer ja huule- ja/või suulaelõhe, väike pea, lampjalgsus, väike lõualuu.
Esimest korda uuris ja sõnastas selle haiguse sümptomeid 1960. aastal dr John Edwards. Ta tuvastas seose teatud sümptomite ilmnemise vahel ja näitas ka üle 130 defekti, mida selle haiguse ajal täheldatakse. Peaaegu kõik need haigusnähud on üsna selgelt märgatavad ja olemasolevate ravimeetodite abil pole neid võimalik kõrvaldada. Võib-olla tulevikus selline võimalus tekib, kuid sellest on veel vara rääkida.
Miks tekib Edwardsi sündroom
Kui "Edwardi sündroomi" diagnoos tehti raseduse ajal, on raseduse katkemise või surnultsündimise tõenäosus äärmiselt suur. Edwardsi sündroom tekib üsna sageli juhuslikult ja kahjuks ei saa seda vältida.
Praegu puuduvad ennetavad meetmed selle kromosoomianomaalia ärahoidmiseks, kuna selle arengu täpsed põhjused pole välja selgitatud, kuid on mõned põhjused, miks lootehaiguse risk võib suureneda. Need sisaldavad:
- ebasoodsad keskkonnategurid;
- kokkupuude kiirguse ja toksiliste kemikaalidega, samuti kiirgusega;
- tubakas ja alkohol;
- pärilikkus – teatud haiguste eelsoodumus võib põlvest põlve edasi anda;
- kokkupuude teatud ravimitega;
- abikaasade sugulus;
- kõige olulisem tegur on lapseootel ema vanus – alates 35. eluaastast suureneb oluliselt tõenäosus, et lootel haigestub Edwardsi sündroom ja mõned muud kromosomaalsed haigused.
Edwardsi sündroomi vormid
Selle kromosomaalse anomaalia tagajärgi mõjutab suuresti embrüo arengustaadium, milles see ilmneb. Edwardsi sündroomi täisvorm on kõige raskem, see areneb tingimusel, et kolm kromosoomi tekkisid ajal, mil oli ainult üks rakk. Järgneva jagamise käigus kantakse lisakoopiad järgmistesse lahtritesse. Sellest tulenevalt täheldatakse igas rakus häiritud kromosoomide komplekti.
Teist Edwardsi sündroomi vormi nimetatakse mosaiigiks, sest sel juhul on terved ja muteerunud rakud nagu mosaiik. See esineb 10% juhtudest. Edwardsi sündroomile omased nähud on sel juhul nõrgemad, kuid normaalne areng on raskendatud. Liigne kromosoom tekib ajal, mil embrüol on juba mitu rakku. Moonutatud geneetiline komplekt sisaldub ainult kehaosas ja ülejäänud rakud on terved. Aeg-ajalt juhtub, et rakud koonduvad organitesse, mida saab eemaldada – sel juhul saab haigust ennetada.
Teine võimalus osalise trisoomia jaoks on võimalik translokatsioon. Sel juhul ei täheldata mitte ainult kromosoomide mittedistantsi, vaid ka nn translokatsiooni ümberkorraldust, mis põhjustab teabe liiasust. Kahe kromosoomi geneetilist järjestust saab osaliselt vahetada. Kui üks neist kromosoomidest on 18., liiguvad selle geenid teise kohta. Edwardsi sündroomi translokatsioonivorm võib ilmneda isegi sugurakkude küpsemise staadiumis või juba embrüo moodustumise ajal. Ühes rakus paikneva translokatsiooni ajal on lisaks 18 kromosoomi paarile ka teises kromosoomis teabe lisakoopia. Sel juhul ei ilmne kõrvalekalded nii olulisel määral, kuna sellel asuvad geenid ei dubleerita.
Edwardsi sündroomi uurijad on jõudnud järeldusele, et 80% haigusjuhtudest esineb trisoomia täisvormis, 10% mosaiikidena. Ülejäänud juhtumeid esindavad haiguse ja häirete translokatsioonivormid, mille tõttu kariotüübis ilmub 2 lisakromosoomi.
Kui levinud on Edwardsi sündroom?
Sellel teabel on erinevates allikates erinev tähendus: alumist piiri võib nimetada väärtuseks 1 10 000-st ja ülemist - 1: 3300 vastsündinut. Praegu on keskmine väärtus 1:7000, mis on peaaegu 10 korda harvem kui Downi sündroomi juhtumid.
Uurimistöö käigus märgiti, et tõenäosus, et vastsündinul tekib lisakromosoom 18, suureneb proportsionaalselt naise vanusega. See kehtib ka muude trisoomiast põhjustatud kõrvalekallete kohta. Väärib märkimist, et Edwardsi sündroomi puhul ei sõltu haiguse esinemissagedus nii palju ema vanusest kui Downi sündroomi ja Patau sündroomi korral, see tähendab vastavalt 13. ja 21. kromosoomi trisoomia korral.
Statistika kohaselt suureneb risk 45 aasta pärast 0,7% -ni, see tähendab, et tõenäosus, et sellise sündroomiga laps sünnib, suureneb mitu korda - kuni 1:140-150. Abikaasade keskmine vanus on naisel 32,5 ja mehel 35 aastat. Oluline on meeles pidada, et esinemise tõenäosus ja Edwardsi sündroomiga haige lapse saamise tõenäosus on erinevad mõisted. Keskmine riskimäär on vastsündinutel 1:7000 ja eostamisel 1:3000 ehk üle 2 korra sagedamini. Kromosoomihäiretega lapse sünd on võimalik nii 30- kui 20-aastaselt, see haigus võib ilmneda terve paari lapsel.
Edwardsi sündroom on anomaalia, millel on otsene seos lapse sooga – poistel on see palju harvem. Uuringute kohaselt esineb Edwardsi sündroomi tüdrukutel 3 korda sagedamini kui poistel. Mõned teadlased oletavad, et sellist statistikat saab seletada X-kromosoomi mõjuga. On võimalus, et lisakromosoomi 18 juuresolekul annab selline kombinatsioon stabiliseeriva efekti ja 18. trisoomiaga isased sügoodid lükatakse organismi poolt tagasi.
Kas testitulemused on alati täpsed?
Kuigi raseduse ajal võib esineda mõningaid sümptomeid, mis võivad olla iseloomulikud Edwardsi sündroomile, ei arene see tegelikult välja nii sageli. Väga oluline on jääda rahulikuks, kui on leitud üksikuid sümptomeid või ebaselgusi. Üsna sageli jagavad naised erinevates foorumites oma kogemusi ja testitulemusi. Peaasi on enne diagnoosi panemist kuulata pädevate spetsialistide arvamust, kes viivad läbi üksikasjalikud uuringud.
Geneetilise testimise tulemuste järgi diagnoositakse sündroom palju täpsemalt kui ultraheli ja vereanalüüside tulemuste järgi, mistõttu ei tasu enne põhjalike uuringute läbiviimist muretseda. Sageli on juhtumeid, kui vereanalüüs näitab, et hormoonide tase ei vasta raseduse ajal normile, kuid samal ajal sünnib terve laps.
Kui klassikaliste testide tulemustes on kahtlusi, määrab arst täiendava uuringu - enamasti ühe invasiivse meetodi. Juhtumeid, kus geenitesti tulemused osutusid valedeks, on üliharva, nende täpsus on üle 99%.
Edwardsi sündroom raseduse ajal
Liigse 18. kromosoomiga loote areng kulgeb teisiti kui normaalses embrüos. See võib mõjutada tiinuse ajastust: üsna sageli sünnivad sellise diagnoosiga sünnijärgsed lapsed - nende rasedusaeg ületab 42 nädalat. Reeglina kulgeb rasedus komplikatsioonidega. On mitmeid märke, mille järgi arstid võivad kahtlustada loote haiguse esinemist.
Üks Edwardsi sündroomiga kaasneda võivaid märke on loote vähene aktiivsus. Eelkõige võib südame löögisagedus (bradükardia) väheneda. Polühüdramnion on üsna tavaline. Kuna Edwardsi sündroomi korral on platsenta suurus tavaliselt aegunud ja väiksem, ei suuda naise keha tagada loote normaalset arengut. Kuid kõik need märgid ei saa olla diagnoosimisel piisavaks aluseks.
Üsna sageli on juhtumeid, kui 2 asemel tekib 1 nabaarter, mille tagajärjel tekib embrüos hapnikupuudus – sel põhjusel kogevad paljud lapsed sündides asfiksiat. Sel juhul võib täheldada kõhuõõne songa (omfalotseeli). Ultraheli diagnostika võimaldab tuvastada veresoonte põimikutest moodustisi - tegelikult on need vedelikuga täidetud õõnsused, mis ei kujuta endast ohtu. Tavaliselt kaovad need 26. nädalaks. Olukorra teeb keeruliseks asjaolu, et üsna sageli kaasnevad selliste moodustistega kõikvõimalikud geneetiliselt määratud haigused. Näiteks Edwardsi sündroomiga leitakse tsüstid peaaegu 30% lastest. Selliste moodustiste leidmisel saadetakse naine geenikonsultatsioonile.
Teine levinud esinemine Edwardsi sündroomi puhul on kehakaalu puudumine (keskmiselt veidi üle 2 kg) ja väljendunud alatoitumus (kroonilised söömis- ja seedehäired).
Seda haigust iseloomustab suur emakasisene suremus – kuni 60% Edwardsi sündroomiga diagnoositud loodetest sureb emakas.
sünnieelne diagnoos
Kuna Edwardsi sündroom on väga raske kromosomaalne häire, on väga oluline haigus õigeaegselt diagnoosida raseduse ajal.
Õigest diagnoosist sõltub kogu patsiendi edasine saatus. Diagnostikat on võimalik ja vajalik läbi viia sünnieelses staadiumis, et võtta vajalikud meetmed või katkestada rasedus õigeaegselt. Loote sünnieelse diagnoosi läbiviimiseks on mitu võimalust.
Ultraheliuuringul võivad ilmneda kaudsed märgid, mis viitavad ainult kromosoomianomaalia võimalusele. Nende hulka kuuluvad mitmesugused sisemised väärarengud, madal loote kaal, suur amnionivedeliku kogus ja mõned teised.
Selleks, et diagnostika tulemused kajastaksid õigesti hetkeseisundit, on väga soovitav, et naine läbiks sünnieelse sõeluuringu. Selle diagnostilise meetodi eesmärk on maksimaalse täpsusega tuvastada loote kromosomaalsete ja muude kõrvalekallete oht. Sellist diagnostikat tehakse kõigile naistele - see võimaldab teil nende hulgas tuvastada kõrge riskiga rühma kuuluvad isikud. Ohu avastamisel annab spetsialist saatekirja invasiivseks testimiseks, mille tulemused kinnitavad või kummutavad kahtlusi.
Sünnieelne sõeluuring jaguneb kaheks etapiks. Esimene langeb 11-13 rasedusnädalale, sel perioodil viiakse läbi biokeemiliste parameetrite uuring. Praegusel ajal on ultraheliga veel raske anomaaliate olemasolu hinnata ning loote arengu kohta saadud teavet ei saa pidada piisavalt täpseks. Lõplikku diagnoosi ei panda, naine ainult langeb või ei kuulu riskirühma. Selles etapis tehakse hormoonide taseme vereanalüüs. Esimesel trimestril ei ole sõeluuringu tulemused veel lõplikud, need viitavad vaid võimalusele, et lapsel on Edwardsi sündroom. Arvutused põhinevad verevalkude analüüsil, mis on usaldusväärsed näitajad loote seisundi kohta raseduse ajal.
Nende valkude hulka kuuluvad plasmas toodetud valk A (PAPP-A) ja β-hCG. See on inimese kooriongonadotropiin (hCG), valk, mida toodavad embrüo membraanide rakud ja seejärel platsenta. Alfa-ühik on hCG ja mõnede teiste hormoonide tavaline näitaja ning nende beeta-subühikud on erinevad. Sel põhjusel määratakse veres hCG β-subühik.
Sünnieelse sõeluuringu teine etapp on täpse teabe saamine loote seisundi kohta. Analüüsiks võetakse koeproov, mis läbib geneetilise uuringu. Proovi võtmiseks võib kasutada mis tahes meetodit. Kõige täpsemad on amniotsentees (lootevee proovi võtmine) ja kordotsentees (nabaväädivere proovi võtmine).
Katsetulemuste põhjal saab hinnata kariotüübi seisundit. Negatiivne tulemus tähendab, et kromosoomianomaaliaid pole tuvastatud, kuid positiivne vastus kinnitab kartusi ja diagnoosi alust.
Invasiivsed testimismeetodid
Kõige täpsemad ja usaldusväärsemad meetodid peetakse invasiivseteks meetoditeks, mis nõuavad kirurgilist sekkumist ja tungimist loote membraani. Need erinevad üksteisest proovide uurimiseks kuluva aja ja loote riskide poolest. Optimaalne proovivõtumeetod määratakse arsti soovituste alusel ja see sõltub suuresti loote arengust ja naise anatoomilistest iseärasustest. Invasiivsete protseduuride läbiviimisel on abordi ja tüsistuste oht. Sellised protseduurid on ette nähtud ainult siis, kui haigusrisk on suurem kui tüsistuste oht.
Juba algstaadiumis on võimalik teha koorioni villuse biopsia (VVK) - alates 8. nädalast. See on CVS-i peamine eelis, sest mida varem on diagnoosi tulemused teada, seda suurem on võimalus tüsistusi vältida. Analüüsiks võetakse emakast proov platsentamembraani ühest kihist, koorionist. Selle struktuur langeb praktiliselt kokku lootekoe struktuuriga. Analüüsi abil on võimalik diagnoosida mitte ainult kromosomaalsete ja geneetiliste haiguste, vaid ka emakasisese infektsiooni risk. Proovi suurus ei tohiks raseduse kulgu oluliselt mõjutada.
Hilisemateks kuupäevadeks, alates 14. nädalast, sobib teine proovivõtumeetod, amniotsentees. Selle olemus seisneb selles, et loote amnionimembraanidesse tungib sond, mille abil võetakse lootevett - need sisaldavad looterakke. Kuna sel juhul on tungimise aste suurem, on erinevate tüsistuste tõenäosus mõnevõrra suurem.
Teine invasiivne meetod on kordotsentees. Soovitatav on seda teha mitte varem kui 20. nädalal. Selle olemus on võtta loote nabaväädivere proov. Seda meetodit muudab keeruliseks asjaolu, et nõel peab sisenema nabanööri anumasse ja ebatäpsused on vastuvõetamatud. Protseduur on järgmine: läbi eesmise kõhuseina torgatakse spetsiaalne punktsiooninõel, millega võetakse väike kogus verd – tavaliselt umbes 5 milliliitrit. Kogu protsessi juhib ultraheliaparaat. Seda meetodit saab kombineerida amniotsenteesiga, kui testitulemustes on kahtlusi.
Kõik need protseduurid ei ole täiesti valutud ja 100% ohutud sündimata lootele, kuid geneetilised haigused, eriti näiteks Edwardsi sündroom, võivad olla palju ohtlikumad. Sel põhjusel soovitatakse naistel tungivalt testida.
Kui materjal on võetud ühel kolmest loetletud meetodist, saadetakse see kontrollimiseks. Spetsialistid viivad läbi geneetilise materjali põhjaliku kromosoomianalüüsi, mille järel on võimalik diagnoosi kinnitada või ümber lükata.
Kuna invasiivsed meetodid hõlmavad tungimist membraanidesse, suureneb lootele kahjulike mõjude oht. Vead nende protseduuride ajal on vastuvõetamatud, kuid võivad tekkida tüsistused. Eelkõige on võimalik tõsiste haiguste ilmnemine ja kaasasündinud väärarengute teke.
Olukorra muudab keeruliseks asjaolu, et mõnel juhul võib pärast selliseid invasiivseid protseduure raseduse spontaanse katkemise oht suureneda. Nii või teisiti peavad tulevased vanemad ise otsustama, kas loote geneetiliste omaduste kohta teabe saamiseks tasub loote raseduse katkemise ohtu seada.
Mitteinvasiivsed testimismeetodid
Sellised meetodid on lootele täiesti ohutud, kuna need ei vaja membraanidesse tungimist, mistõttu puudub otsene mõju sündimata lapsele. Riski määramise täpsus ei ole madalam kui invasiivsete meetodite kasutamisel. Näiteks kui karüotüpiseerimiseks on vaja ema vereproovi, kuna see sisaldab vaba loote DNA-d, ekstraheerivad spetsialistid need, seejärel kopeerivad ja analüüsivad. See meetod võimaldab teil üsna suure täpsusega tuvastada ebanormaalsete kromosomaalsete muutuste olemasolu.
Sünnitusjärgne diagnoos
Kuna Edwardsi sündroomi iseloomustab üsna suur hulk väljendunud kõrvalekaldeid, on seda üsna lihtne diagnoosida isegi väliste ilmingute abil. Sellest aga ei piisa ning vaja on protseduur, mille põhjal saab diagnoosi panna.
Võite kasutada täiendavaid uurimismeetodeid, sealhulgas:
- Ultraheli, kuna patoloogiate tuvastamiseks on vaja uurida siseorganeid, sealhulgas südant;
- aju tomograafia, mis võib paljastada kõrvalekaldeid;
- konsultatsioonid lastearstidega: silmaarst, endokrinoloog, neuroloog, otolaringoloog; oluline on leida spetsialistid, kes mõistavad seda sündroomi ja on töötanud patsientidega, kellel on see sündroom diagnoositud.
- Et teada saada, kas haiguse kirurgiline ravi on võimalik, on vajalik kvalifitseeritud kirurgi konsultatsioon.
Diagnoosi kinnitamiseks (või ümberlükkamiseks) on oluline hankida kogu vajalik geneetiline informatsioon. Seda saab saada karüotüpiseerimise protseduuriga, mille käigus analüüsitakse geneetilist koodi.
Karüotüpiseerimine on kromosoomikomplekti analüüs, mis seisneb vanemate venoosse vere analüüsis. Pärast analüüsiks vajalike rakkude saamist asetatakse need inkubaatorisse ja kopeeritakse. Kui saadakse piisav arv rakke, peatatakse nende jagunemine, need värvitakse ja uuritakse raku tuumas olevaid kromosoome.
Analüüs nõuab suuremat tähelepanu ja mõningaid ettevalmistavaid protseduure, seega saavad tulemused teada umbes 2 nädala pärast.
Kui täpsed on geneetilise testimise tulemused?
Kõik loetletud geneetilise testimise meetodid on väga täpsed, eriti võrreldes teiste analüüsimeetoditega. Tulemuse täpsus võib ületada 90%, samas kui valevastuse tõenäosus jääb üsna väikeseks.
Mosaiikvormi puhul on haiguse avastamise tõenäosus väiksem, kuna pole võimalik jälgida, millised rakud satuvad uuritavasse materjali. See on tulemuste kindlaksmääramisel kõige raskem. Seega, kui analüüsiks võeti ainult terved rakud, on mosaiikvormi tuvastamine võimatu. Ja vastupidi: proovi võivad sattuda häiritud kromosoomikoostisega rakud. Siis on põhjust positiivseks vastuseks, kuid teave ei ole piisavalt täielik.
Igal juhul on parem kontrollida saadud andmeid muude meetoditega - ultrahelist kuni otsese sünnitusjärgse uuringuni.
On väga oluline, et vanemad mõistaksid, et kui lapsel avastatakse testimise tulemusena geneetiline anomaalia, siis see jääb eluks ajaks. Rakud ei saa kariotüüpi muuta ja lähituleviku kohta selliseid ennustusi pole.
Kui kaua elavad Edwardsi sündroomiga lapsed?
Trisoomia 18 põhjustatud häired on palju tõsisemad kui 21. trisoomia (st Downi sündroomi) põhjustatud häired. Downi sündroomiga võivad inimesed elada aastakümneid, nad kohanevad mingil määral sotsiaalse eluga.
Edwardsi sündroomiga lapse eluiga on äärmiselt lühike: enamik lapsi ei ela aastani, vaid 10% lastest elab selle vanuseni. Ligikaudu 50% patsientidest sureb esimese 2 kuu jooksul, samas on seos sooga. Selle sündroomiga poisid elavad umbes 60 päeva ja tüdrukud umbes 280 päeva.
Edwardsi sündroomi välised kõrvalekalded
Haiguse välised ja sisemised ilmingud võivad oluliselt erineda sõltuvalt loote arengu omadustest. Kõige sagedamini ilmnevad kromosomaalsed häired embrüo arengu algfaasis, seega mõjutavad need kogu organismi arengut. On mitmeid väliseid märke, mis suure tõenäosusega viitavad Edwardsi sündroomi esinemisele vastsündinutel.
Selle haiguse üheks iseloomulikumaks tunnuseks on koljuluude moonutatud kuju: kolju on pea ülaosast lõuani välja piklik, diagnoositakse aga mikrotsefaalia (kolju ja aju suuruse vähenemine) või sageli tehakse hüdrotsefaalia (vedeliku kogunemine ajus). Otsmik on kitsas ja kuklakühm laiem ja silmatorkavam, kõrvad asetsevad madalamal kui normaalses arengus. Lõualuud on deformeerunud - sageli põhjustab see alalõua olulist vähenemist, see muutub kitsaks ja vähearenenud. Selle tagajärjel on suu ka ülahuule lühenemise tõttu väike ja sageli kolmnurkne. Taevas on kõrgel, kohati on tühimik. Kael võib olla lühenenud, iseloomuliku voldiga.
Palpebraalsed lõhed on kitsamad ja lühemad kui vaja, ninasild on laienenud ja surutud - see on eriti märgatav hoolimata sellest, et nina on tavaliselt kitsendatud, nina luud võivad visuaalselt puududa. Silmamuna alluvad ka muutused ja häired, mis põhjustavad katarakti ja koloboomi, st osa silmamembraani puudumist. Lisaks võib esineda muid nägemiskahjustusi.
Kõrvad on madala asetusega ja deformeerunud, sageli horisontaaltasapinnas. Sageli puudub kõrvasagar ja mõnikord puudub ka tragus. Väliskuulmine on sageli ahenenud, mõnikord võib see täielikult puududa.
Luu luustikuga on seotud lai valik häireid. Esiteks ei saa liigesed korralikult funktsioneerida, mistõttu jalad ja käed ei saa painduda ega venitada nii nagu peaks. Lisaks on jalad vähearenenud, seetõttu muutub nende kuju, nad muutuvad vähem liikuvaks. Pöial lüheneb ning teine ja kolmas kasvavad kokku, vahel nii tugevalt, vahel tekivad lestataolised jäsemed. 80% juhtudest moodustub longus kaarega jalg, väljaulatuv kannaosa ja lühike pöial.
Puusaliigese liigse liikuvuse tõttu tekivad sageli nihestused.
Sõrmede padjanditel võib kaarte olla 10 korda rohkem kui tavaliselt, kuid sõrmedel ei ole paindevolti. Ligi 30% patsientidest tekivad peopesadele põikivaod ja palju kammkarpe.
Muuhulgas on Edwardsi sündroomiga rindkere kuju deformeerunud - see laieneb ja roietevahelised ruumid vähenevad, mistõttu see muutub lühemaks ja laiemaks.
Toimuvad olulised muutused ja siseorganid. Peaaegu kõigil patsientidel on südamehaigus. Reeglina iseloomustab seda arterite ja aordi ventiilide ebapiisav areng. Sel juhul on üsna sageli interventrikulaarse vaheseina defekt.
Ainevahetusprotsessides, näiteks endokriinsüsteemi töös, on väga tõsised häired. Kromosomaalsete häirete tõttu ei saa näärmed normaalselt funktsioneerida, mistõttu kasv aeglustub oluliselt. Hormonaalsed häired põhjustavad nahaaluse koe vähest arengut. Igal kümnendal on neerupealiste või kilpnäärme talitlushäire.
Vähenenud lihastoonus tavaliselt aja jooksul tõuseb ja vereringe paraneb.
Ligikaudu pooltel patsientidest täheldatakse soolestiku ebanormaalset arengut. Kõige sagedamini peitub see anomaalia selle ebaharilikus asukohas, samal ajal kui ilmub sooleseina kihtidest moodustunud kott ja söögitoru kitseneb liiga järsult. Neerud on sageli segmenteeritud või ebaregulaarselt kaarduvad ning võib esineda kusejuhade dubleerimist.
Muutused mõjutavad ka suguelundeid. Poistel ei pruugi munand laskuda munandikotti (krüptorhidism) ja peenise struktuur muutub. Tüdrukutel moodustub hüpertrofeerunud kliitor ja munasarjad on vähearenenud.
Üldiselt on Edwardsi sündroomi väliste ja sisemiste kõrvalekallete pilt järgmine. 100% juhtudest täheldatakse kolju struktuuri kõrvalekaldeid ja näo kuju muutumist. Peaaegu 97%-l esineb lõualuu langus (mikrogeenia), veidi enam kui 95%-l juhtudest on kõrvade ehitus ja paiknemine häiritud. Peaaegu 90%-l patsientidest täheldatakse kolju pikenemist, 78%-l kõrget suulaetunnet ja 71%-l juhtudest suu vähenemist.
Mis puudutab jäsemete häireid, siis neid esineb 98% patsientidest. Kõige sagedasemad muutused käte (üle 91%) ja jalgade (76%) kujus.
Kardiovaskulaarsüsteemi areng on häiritud enam kui 90% patsientidest. Umbes 1/3 patsientidest esineb urogenitaalsüsteemi häireid ja 55% seedesüsteemi.
Edwardsi sündroomiga imikute toitmine
Kuna arenguanomaaliad on spetsiifilised ja üsna tõsised, on Edwardsi sündroomiga laste toitmine väga keeruline. Kõige tõsisemad probleemid on põhjustatud imemis- ja neelamisrefleksi puudumisest või rikkumisest. Laps kas ei saa piima juua või võib neelamisel lämbuda.
Vaimse arengu kõrvalekalded
Kõigil patsientidel on aju vähearenenud, eriti kehakeha ja väikeaju. See toob paratamatult kaasa häireid vaimses arengus. Kui laps jääb ellu, ilmneb aja jooksul märgatav mahajäämus eakaaslastest.
Reeglina areneb Edwardsi sündroomi täisvormiga oligofreenia keerulisel määral. Haiguse mosaiikvormiga ei pruugi see nii selgelt avalduda. Sageli tekib patsientidel kramplik sündroom (aju düsfunktsioon, millega kaasnevad tahtmatud lihaste kokkutõmbed).
Sündroomi mosaiikvormi tagajärjed ei ole nii tõsised, kuid rikkumised on endiselt arvukad ja märgatavad. Samal ajal ei sõltu haiguse vormi tõsidus tervete ja muteerunud rakkude suhtest. Teine raskus seisneb haiguse vormide mitmekesisuses.
Edwardsi sündroomiga lapse elu nõuab suuremat tähelepanu ja kontrolli. Vaimsete võimete areng kulgeb komplikatsioonidega, suhtlemisel on raskusi. Enamasti suudavad nad mugavust ära tunda ja sellele reageerida, nad saavad õppida naeratama. Kui laps õpib mõnda inimest ära tundma, siis on mõnel juhul võimalik väga piiratud suhtlus. Nõuetekohase hoolduse korral saab laps õppida oma pead tõstma ja sööma.
Kas Edwardsi sündroomi saab ravida?
Olukorra teeb keeruliseks asjaolu, et Edwardsi sündroom ilmneb geneetiliste häirete tagajärjel, mis võivad keharakke erineval määral mõjutada. Selgub, et täielikuks ravimiseks on vaja kõigis haigetes rakkudes olev materjal "parandada". Hetkel teadusavastused sellist protseduuri teha ei võimalda, seega on geneetilised haigused endiselt ravimatud. Eksperdid ei välista, et selline võimalus tekib tulevikus. Hetkel on võimalik siluda vaid patoloogiliste muutuste tagajärgi.
Kuna haigust ei saa täielikult välja ravida, piirdub ravi tavaliselt toetavate meetmetega. Eelkõige püüavad arstid võimalikult palju tõsta patsiendi ja tema pere moraali. Isegi kui võetud meetmed on võimalikult tõhusad, ei ületa tõenäosus, et laps elab vähemalt aasta, 5-10%. Ellujäänud lastel on suur hulk erinevaid kõrvalekaldeid ja häireid.
Sünnil Edwardsi sündroomiga diagnoositud laste arengut iseloomustab alati kõrvalekallete esinemine. Arsti jaoks on suurimaks raskuseks haiguse vormi kindlaksmääramine ja ravi valimine. Väliseid muutusi saab korrigeerida operatsiooniga, kuid varase suremuse tõttu on sellised meetmed enamasti põhjendamatud.
Väliseid häireid saaks korrigeerida operatsiooniga, kuid riskid suurenevad, kuna südame-veresoonkonna süsteemi talitlushäired põhjustavad tüsistusi. Tõsised närvi- ja lihassüsteemi häired on põhjuseks, miks lihasluukonna süsteem ei saa normaalselt areneda – see toob kaasa skolioosi, strabismuse ja isegi lihaste atroofia.
Edwardsi sündroomiga patsientidel on kõhukelme seinte madal toon ja soole atoonia; koos näo luude moonutamisega raskendab see kõik oluliselt rinnaga toitmist. Teatud paranemist võivad põhjustada piimasegud, vahueemaldajad ja lahtistid.
Edwardsi sündroomiga patsientidel on risk haigestuda neeruvähki, mistõttu on vajalik regulaarsed ultraheliuuringud. Urogenitaalsüsteemi ebaõigest toimimisest võivad tekkida tüsistused. Suure tõenäosusega on kõrvapõletik, sinusiit, konjunktiviit, kopsupõletik ja hulk muid haigusi.
Kuna sellise diagnoosiga ilmneb üsna palju rikkeid, on oluline patsiendi seisundit kogu aeg jälgida, et neid õigel ajal märgata ja ravi alustada.
Kas Edwardsi sündroom on pärilik?
Edwardsi sündroomiga kaasneb suur hulk tõsiseid arenguhäireid, mistõttu tekib küsimus, kas see haigus ja kalduvus sellele on päritav? Vastus peitub haiguse põhjuses.
Arvukate kõrvalekallete teke on lisakromosoomide ilmumise tagajärg: 18. kromosoom tekib kas sugurakus või embrüo arengu käigus. See tähendab, et vanemad ise on terved ning nende geneetilises materjalis pole eeldusi Edwardsi sündroomi tekkeks.
Veel üks aktuaalne küsimus: kas muudetud kromosoomikomplekti on võimalik järgmistele põlvkondadele üle kanda? Vastus sellele on eitav, kuid peamiselt ainult seetõttu, et enamik patsiente ei ela reproduktiivse vanuseni. Lisaks on täiendava kromosoomi ülekandmine võimatu isegi teoreetiliselt, kuna suguelundid on vähearenenud ja paljunemisvõimed on täiesti välja arenemata.
Nende faktide ja ka arvukate meditsiiniliste uuringute põhjal võib kindlalt järeldada, et Edwardsi sündroom ei ole päritav.
Kas järgmisel lapsel võib tekkida Edwardsi sündroom?
Kui perre on kunagi sündinud Edwardsi sündroomiga lapsi, siis on üsna loogiline, et abikaasadel tekib küsimus sellise anomaalia kordumise kohta. Teadlased väidavad, et selline nähtus on ebatõenäoline. Selline kõrvalekalle iseenesest on üsna haruldane - see esineb umbes 1% juhtudest. Samas on tõenäosus, et Edwardsi sündroom järgmise raseduse ajal uuesti diagnoositakse, umbes 0,01%.
Mutatsioonide oht sugurakkudes või sugurakkudes võib sõltuda mõnest agressiivsest tegurist. Nende hulka kuuluvad alkoholi tarbimine, kokkupuude sigaretisuitsuga jne. On oluline, et naine jälgiks hoolikalt oma tervist ja hoiduks loote arengut nii otseselt kui kaudselt mõjutada võivate tegurite negatiivsest mõjust. Suur tähtsus ei ole mitte ainult rasedusperiood, vaid ka periood enne rasestumist.
(18. kromosoomi trisoomia) on Downi sündroomi järel levinuim kromosomaalne häire. Edwardsi sündroomi esinemissagedus on 1:5000-1:7000 vastsündinut. Edwardsi sündroomiga tüdrukud sünnivad kolm korda sagedamini kui poisid.
| 1. pilt. Näide Edwardsi sündroomi diagnoosimisest CF-PCR abil. | Joonis 2. Haige laps Edwardsi sündroomiga. |
| Näide Edwardsi sündroomi (18. kromosoomi trisoomia) diagnoosimisest. Lillaga on esile tõstetud markerid, mis asuvad 18. kromosoomi piirkonnas, mis on Edwardsi sündroomi tekkeks kriitilise tähtsusega. Uuritava proovi genotüübis on D18S978 markeril 1 piik (marker ei ole informatiivne), markeritel D18S535 ja D18S386 - 3 piiki (trisoomia), markeritel D18S390 ja D18S819 - annuse mõju - ebavõrdne. kahe tipu kõrguste suhe (trisoomia). Seega tuvastati nelja markeri (D18S535, D18S386, D18S390 ja D18S819) puhul trisoomia, mis võimaldab määrata Edwardsi sündroomi diagnoosi. Geneetiline sugu vastab meessoost - markerite Amelogenin, 4SH, ZFXY, TAFL jaoks on Y-kromosoomile vastavad piigid ja SRY geeni tipp. |
|

"Kuldstandard" kromosomaalsete häirete tuvastamisel kogu maailmas on pikka aega olnud ja on ka edaspidi kromosoomide diferentsiaalse värvimisega karüotüpiseerimise meetod. See meetod võimaldab analüüsida karüotüüpi tervikuna ja määrata suuri (vähemalt 5–10 miljonit aluspaari) kromosomaalseid ümberkorraldusi. Sellel on aga mitmeid piiranguid, nagu töömahukus, kestus (1-2 nädalat), kõrged nõuded uuringut läbiviiva spetsialisti kvalifikatsioonile ja kogemustele ning mõnel juhul tehnilised probleemid (ebapiisav kogus ja kvaliteet). uuritav materjal, mitooside puudumine või kultuuri kasv).
Kvantitatiivse fluorestseeruva polümeraasi ahelreaktsiooni (QF-PCR) meetod, mida üha enam kasutatakse aneuploidiate, sealhulgas Edwardsi sündroomi diagnoosimiseks, on nendest puudustest ilma jäänud (joonis 1). Selle meetodi usaldusväärsus on võrreldav standardse karüotüpiseerimisega, see on kiirem, odavam, materjali kvantiteedi ja kvaliteedi suhtes vähem nõudlik (kuna see ei ole seotud rakukultuuri kasvuga) ning võimaldab üheaegselt analüüsida suurt hulka proove. . Kuid CF-PCR meetodil on ka piirangud: mosaiikjuhtudel suudab see tuvastada ainult kõrgetasemelist mosaiiksust (alates 20%), lisaks ei saa see välistada haruldasemate kromosomaalsete häirete esinemist, mis võivad olla seotud loote väärarengutega. Edwardsi sündroomi sünnieelse diagnoosimise läbiviimisel on lisaks lootematerjalile vaja anda emale ka bioloogilist materjali, et välistada lootematerjali ebaõige proovivõtmise tõttu vale negatiivse tulemuse saamise võimalus. Lootematerjali analüüs tehakse kolme tööpäeva jooksul.
Edwardsi sündroomi korral on sünnieelses arengus väljendunud viivitus, lapsed sünnivad sünnieelse alatoitumusega (keskmine kehakaal sünnihetkel on 2340 g). Edwardsi sündroomi välised ilmingud on mitmekesised (joonis 2). Kõige tüüpilisemad on hilinenud psühhomotoorne areng, skeletilihaste ja nahaaluse rasvkoe hüpoplaasia, kaasasündinud südamerikked, näo ja kolju struktuuri anomaaliad (dolichotsefaalia, mikroftalmia, silmalõhede lühenemine, kõrvade madal asend, mikrognaatia, kaldus lõug), käte ja jalgade hulgi deformatsioonid, seedetrakti, urogenitaalsüsteemi ja kesknärvisüsteemi arengu anomaaliad (lülisamba song, jämesoole ja väikeaju hüpoplaasia). Laste oodatav eluiga väheneb järsult: 90% neist sureb enne aastat kaasasündinud väärarengute tõttu tekkinud tüsistustesse (lämbumine, kopsupõletik, soolesulgus, kardiovaskulaarne puudulikkus).
Edwardsi sündroomi väljakujunemise põhjuseks on 18. kromosoomi kolmekordistumine. Trisoomia 18. kromosoomil on aneuploidsuse erijuht – kromosoomide komplekti olemasolu genoomis, mis erineb selle liigi standardist, mitte aga mitmekordne. . Trisoomia 18 on tavaliselt põhjustatud kromosoomide mittelahkumisest vanema sugurakkude (munarakkude ja spermatosoidide) moodustumisel, mille tulemusena saab laps emalt või isalt täiendava 18. kromosoomi. Sel juhul kannavad anomaaliat kõik lapse keharakud. Kui embrüo mis tahes raku jagunemisel toimub kromosoomide mitteeraldamine, täheldatakse Edwardsi sündroomi mosaiikvarianti (10% juhtudest).
Edwardsi sündroomiga laste saamise risk erinevatel kirjandusandmetel ei muutu või suureneb veidi koos raseda vanusega.
Edwardsi sündroomi sünnieelne diagnoos hõlmab kahte etappi. Esimeses etapis, 11-13 rasedusnädalal, viiakse läbi sõeluuring, mis põhineb peamiselt biokeemilistel parameetritel, kuna varases staadiumis ei võimalda ultraheli Edwardsi sündroomi korral suuri arenguanomaaliaid tuvastada. mida saab tuvastada alles 20-24 nädala pärast. Teatud valkude taseme biokeemiline analüüs raseda naise veres (inimese koorionihormooni vaba β-subühik (β-hCG) ja rasedusega seotud plasmavalk A (rasedusega seotud plasmavalk-A, PAPP-A)), võttes arvesse tema vanust, võimaldab teil arvutada tema jaoks haige lapse saamise riski. Need meetodid ei võimalda aga täpset diagnoosi panna ning sõeluuringu tulemusena moodustub rasedate riskirühm, kellel on suurem tõenäosus sünnitada Edwardsi sündroomiga patsient. Teises etapis viiakse riskirühmas läbi invasiivne protseduur loote seisundi täpseks määramiseks vajaliku lootematerjali saamiseks. Olenevalt gestatsiooni vanusest võib selleks olla koorioni villuse biopsia (8.-12. nädal), amniotsentees (14.-18. nädal) või kordotsentees (pärast 20. nädalat). Saadud loote koeproovides määratakse kromosoomikomplekt.
Molekulaargeneetika keskus teostab Edwardsi sündroomi (sh prenataalset) diagnostikat CF-PCR abil.